Download as PDF, PPTX










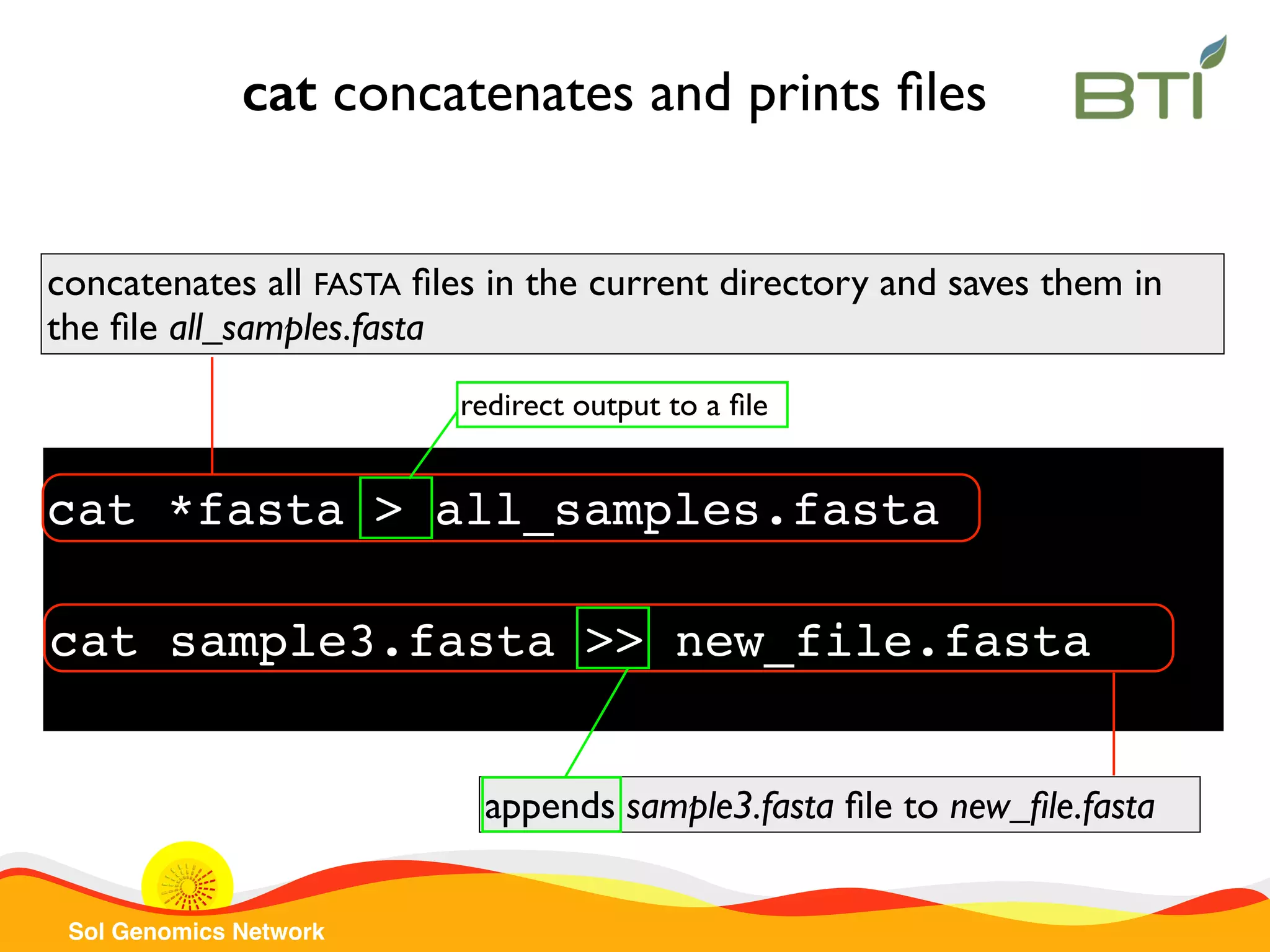
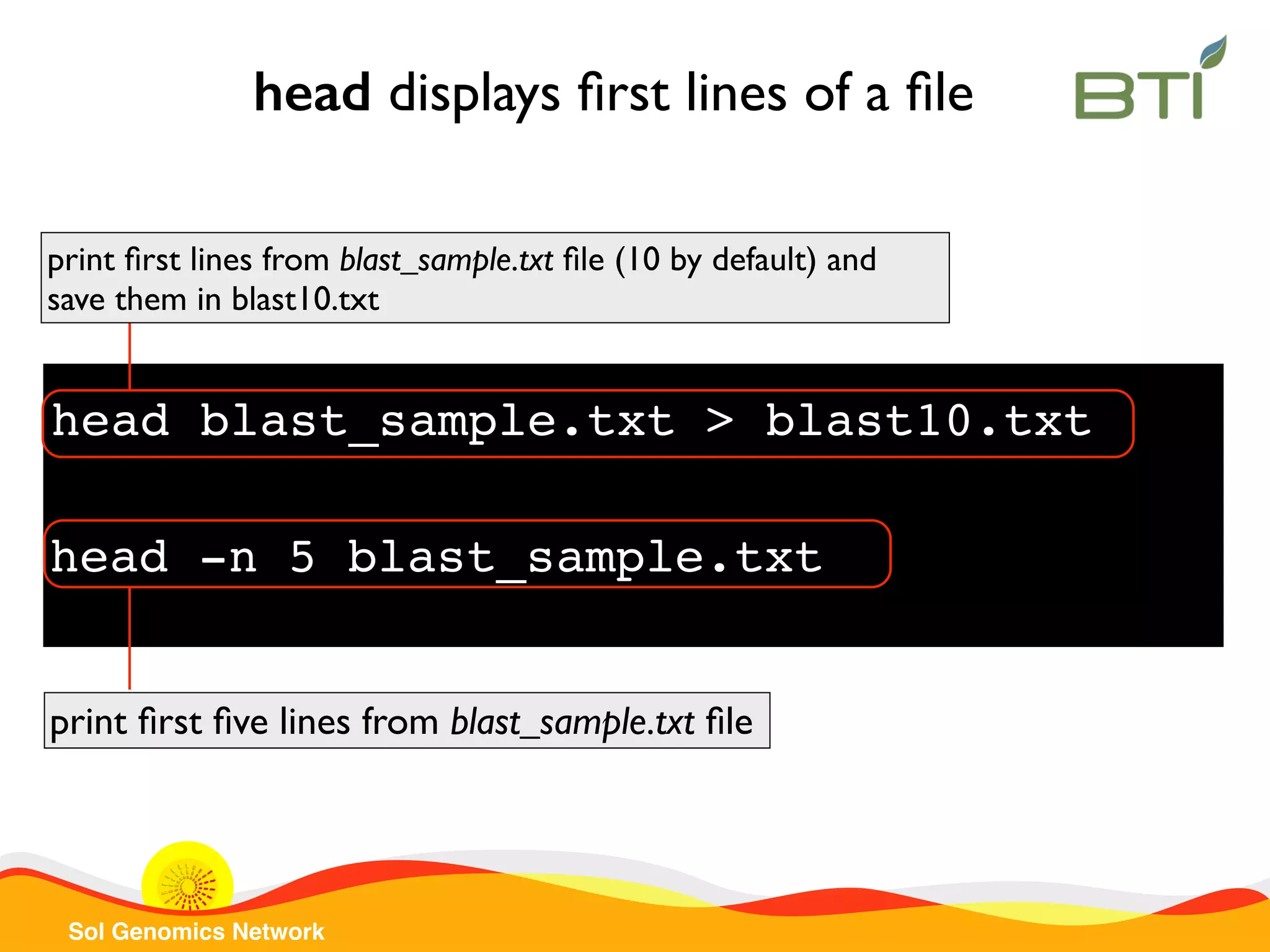







![Sol Genomics Network sed replaces a pattern sed ‘s/A/a/g’ col1.txt replaces all “A” characters by “a” in col1.txt file sed ‘s/Atha/SGN/’ col1.txt replaces Atha by SGN in col1.txt file sed -r ‘s/^([A-Za-z]+)|(.+)/gene 2 from 1/’ col2.txt get species and gene name from col2.txt and print each line in a different format Saves species name in 1 Saves gene name in 2](https://image.slidesharecdn.com/sgnintroductiontounixcommand-line20152-151112200330-lva1-app6891/75/SGN-Introduction-to-UNIX-Command-line-2015-part-2-20-2048.jpg)



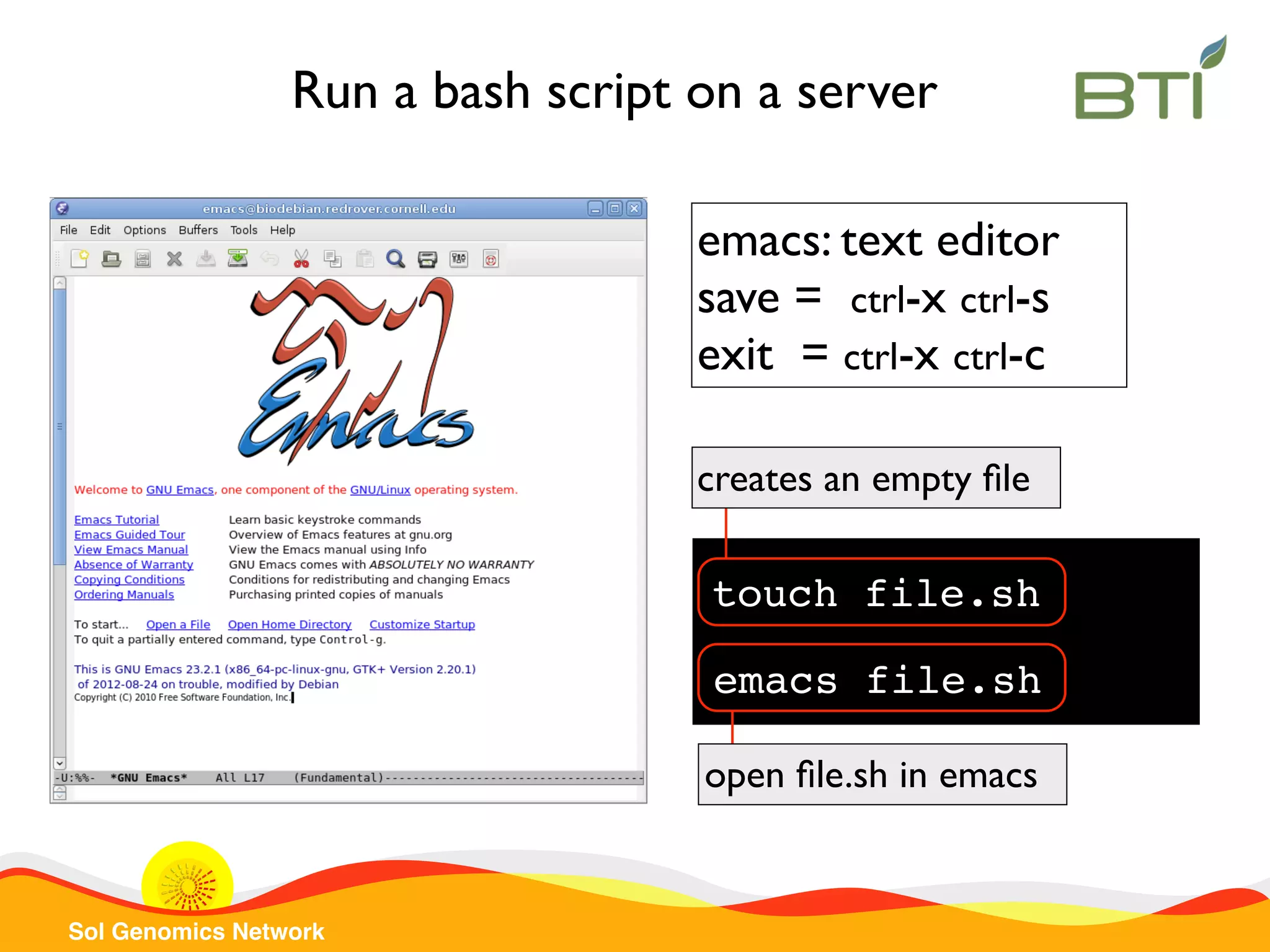




The document is a comprehensive guide on UNIX command-line operations, particularly focusing on file system navigation, text manipulation, file permissions, and command-line pipelines. It details various commands and their usage for handling text files, including commands like 'cat', 'grep', 'sort', and methods for file compression and networking. Additionally, it includes examples and exercises related to bioinformatics tasks involving FASTA and FASTQ file formats, as well as instruction on creating and executing bash scripts.