Download as PDF, PPTX



![16th December 2015 Dimensions of Big Data The 7 Vs of Genomic Big Data • Volume is defined in terms of the physical volume of the data that need to be online, like giga-byte (10 9 ), tera-byte (10 12 ), peta-byte (10 15 ) or exa-byte (10 18 ) or even beyond. • Velocity is about the data-retrieval time or the time taken to service a request. Velocity is also measured through the rate of change of the data volume. • Variety relates to heterogeneous types of data like text, structured, unstructured, video, audio etcetera. • Veracity is another dimension to measure data reliability - the ability of an organization to trust the data and be able to confidently use it to make crucial decisions. • Vexing covers the effectiveness of the algorithm. The algorithm needs to be designed to ensure that data processing time is close to linear and the algorithm does not have any bias; irrespective of the volume of the data, the algorithm is able to process the data in reasonable time. • Variability is the scale of data. Data in biology is multi-scale, ranging from sub-atomic ions at picometers, macro-molecules, cells, tissues and finally to a population [9] at thousands of kilometers. • Value is the final actionable insight or the functional knowledge. The same mutation in a gene may have a different effect depending on the population or the environmental factors.](https://image.slidesharecdn.com/bda2015-tutorial-part2-datadatabases-160311113748/75/Bda2015-tutorial-part2-data-amp-databases-7-2048.jpg)










![PRECISE RESULTS MLH1[Gene Name] AND Human[Organism]](https://image.slidesharecdn.com/bda2015-tutorial-part2-datadatabases-160311113748/75/Bda2015-tutorial-part2-data-amp-databases-41-2048.jpg)









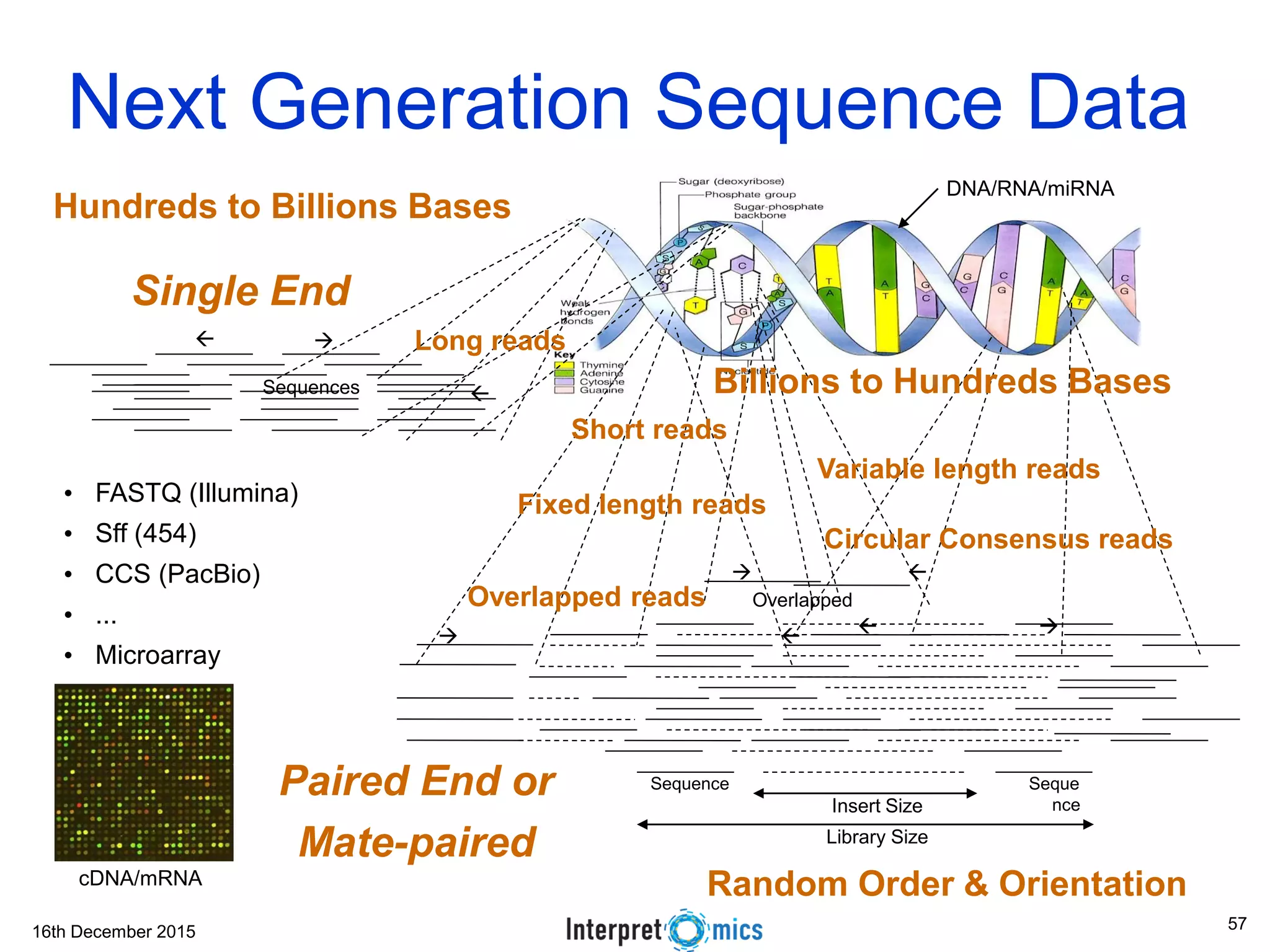
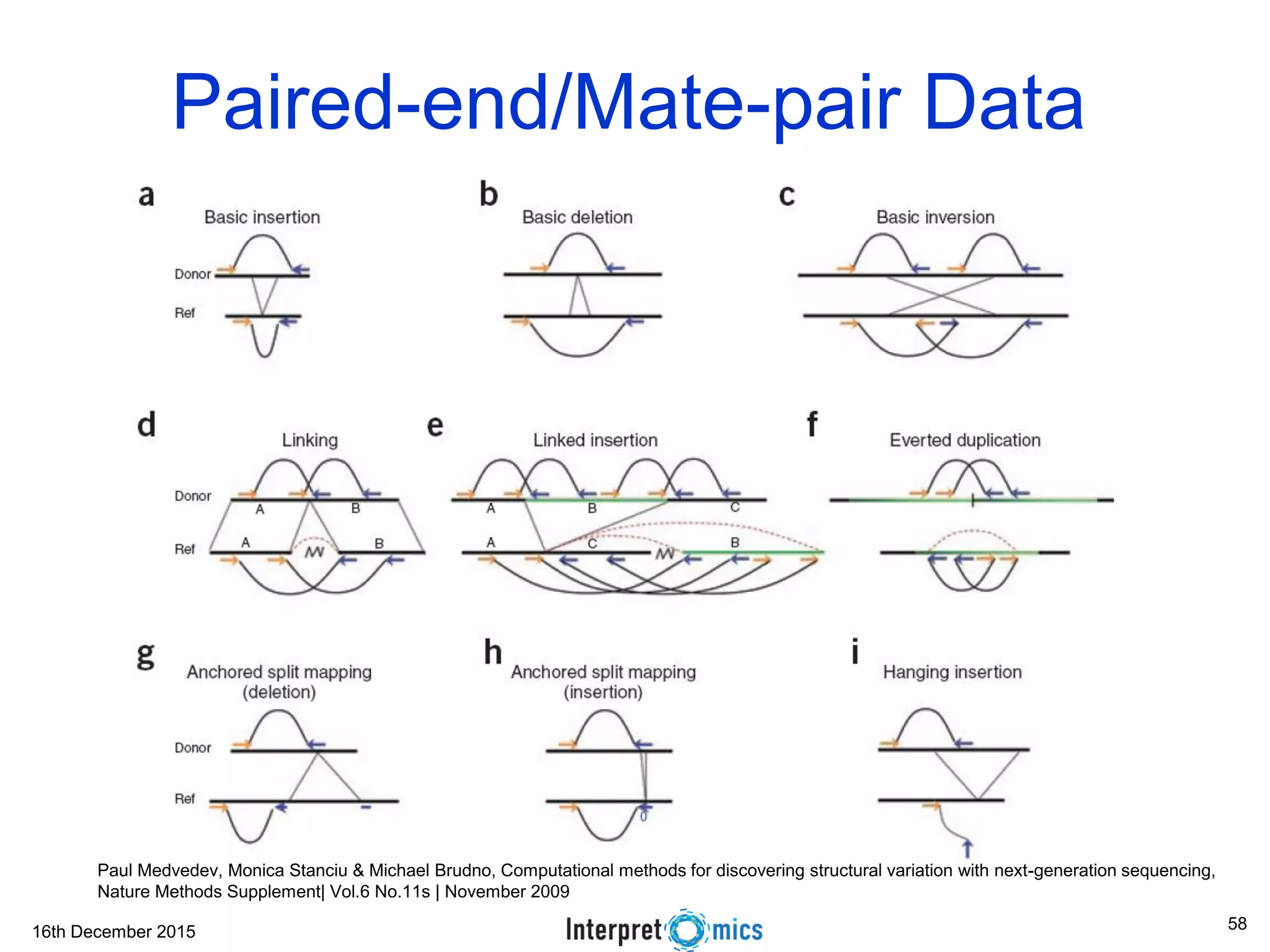


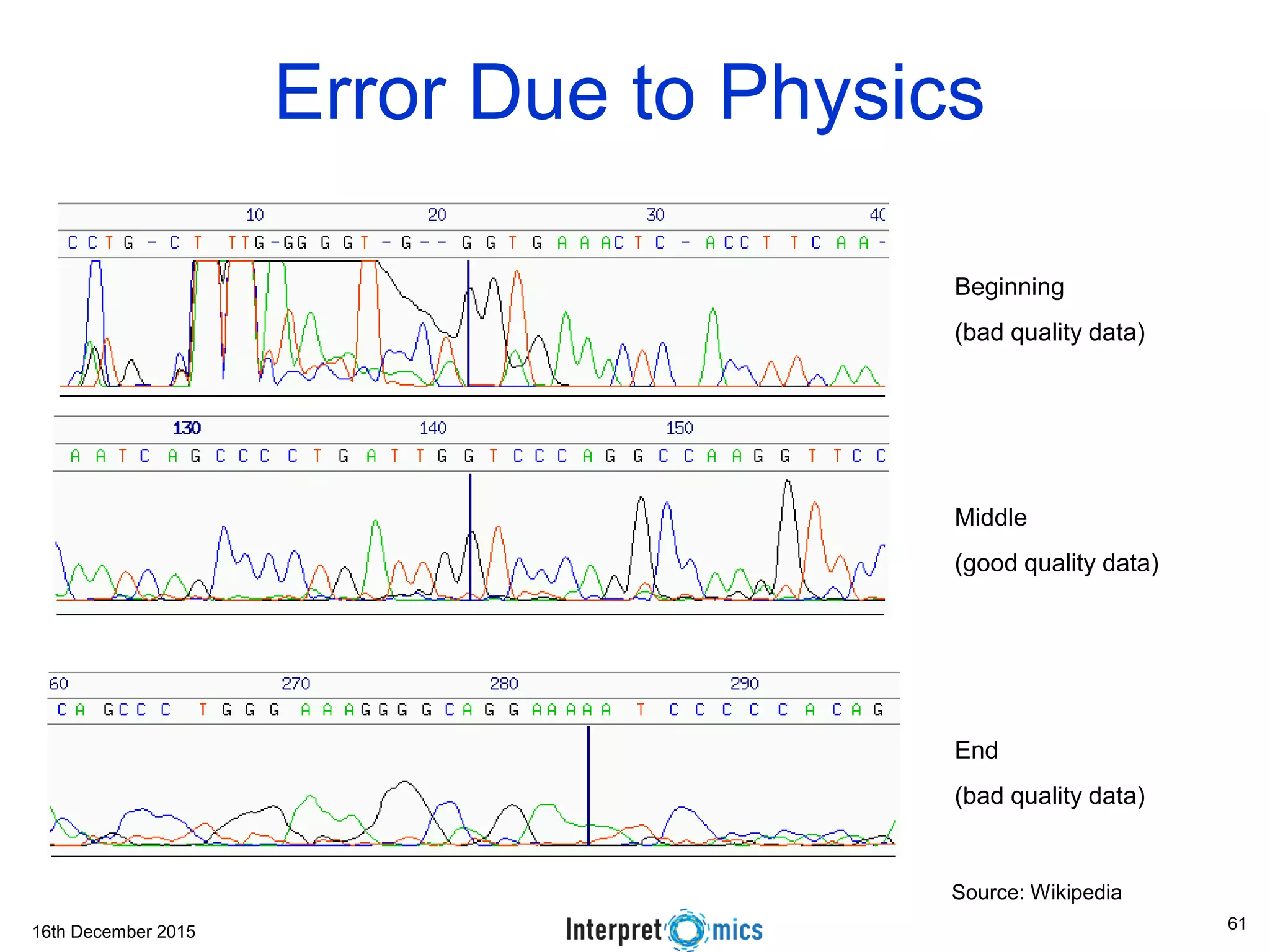



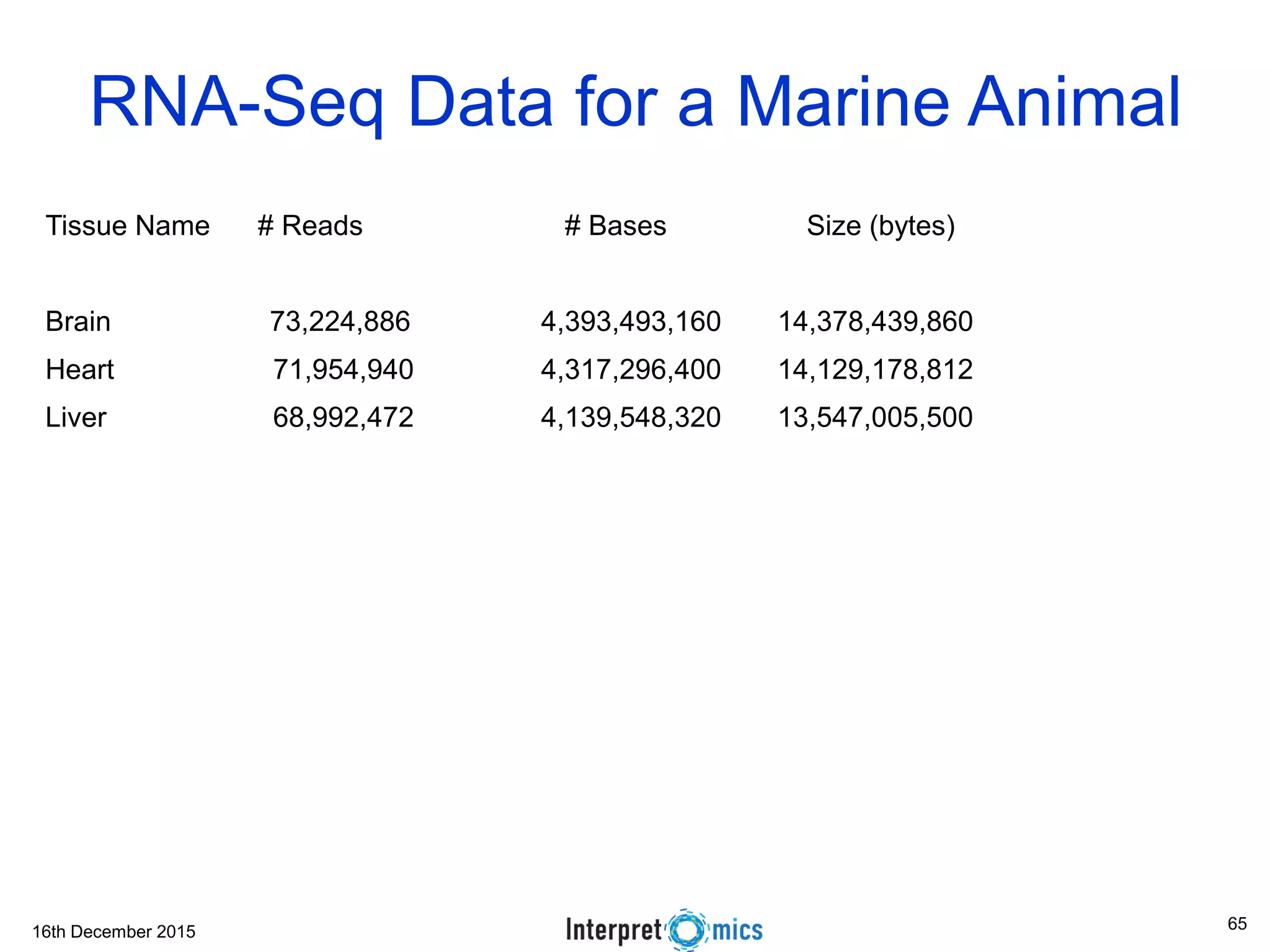


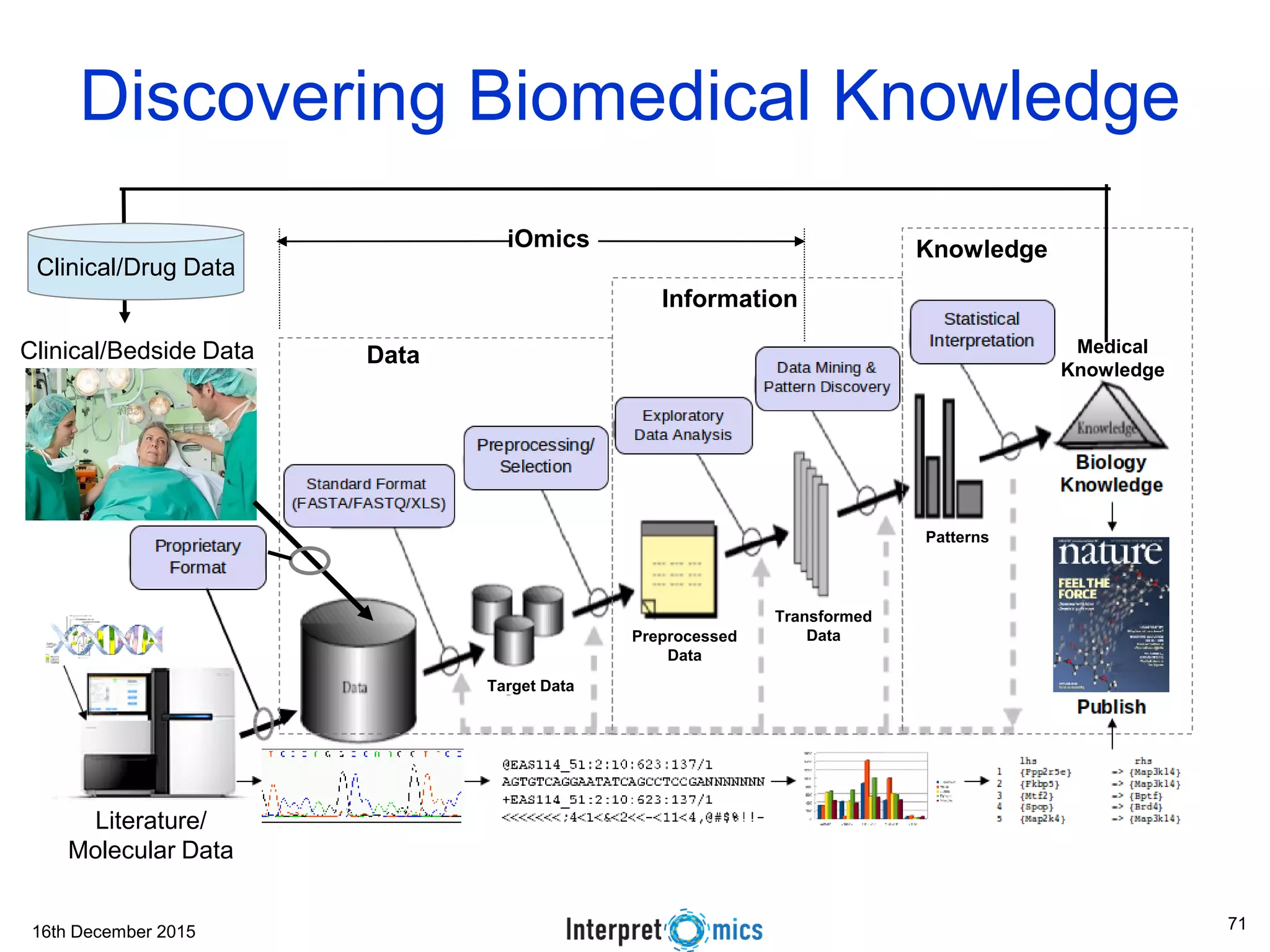
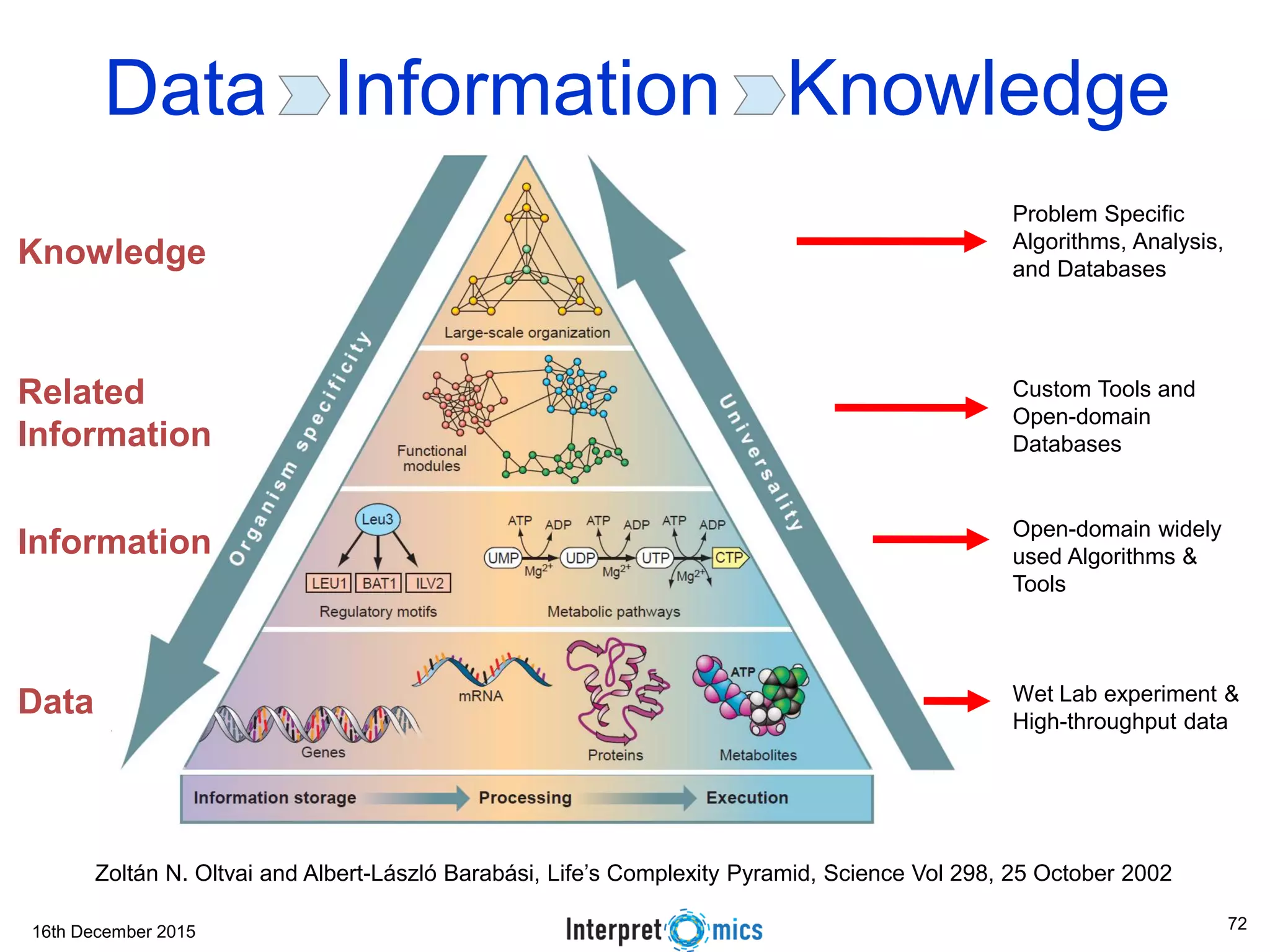
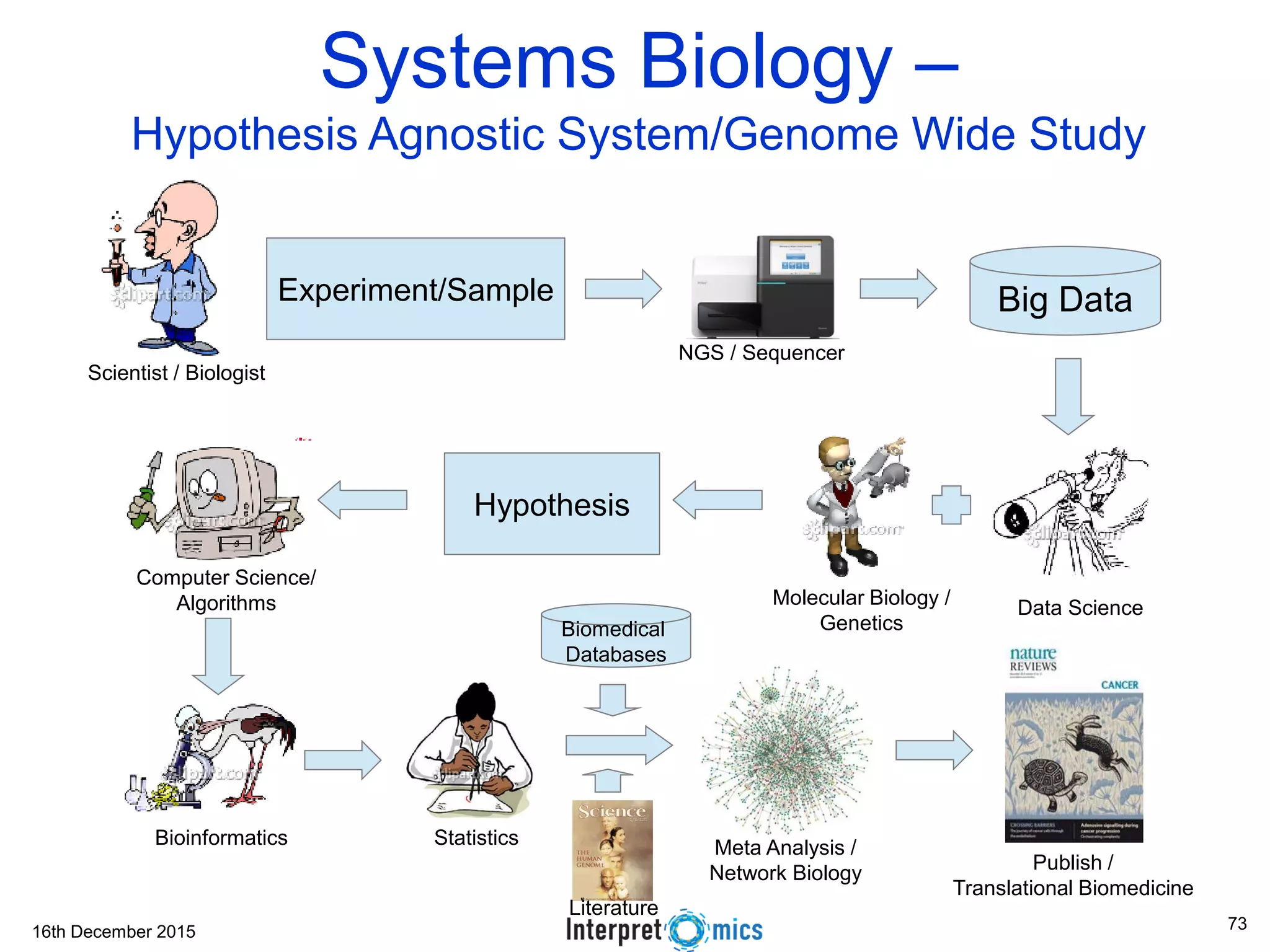



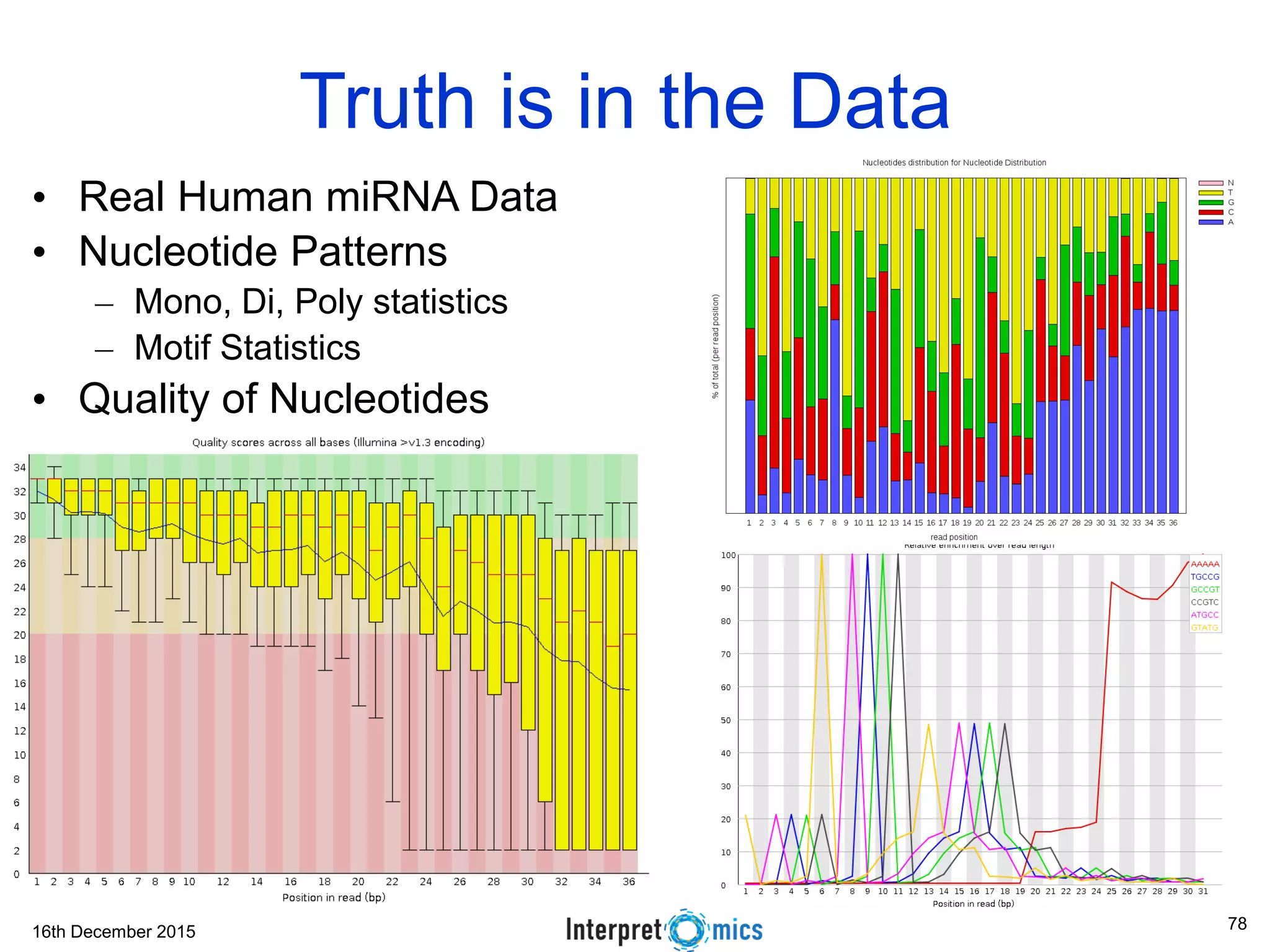
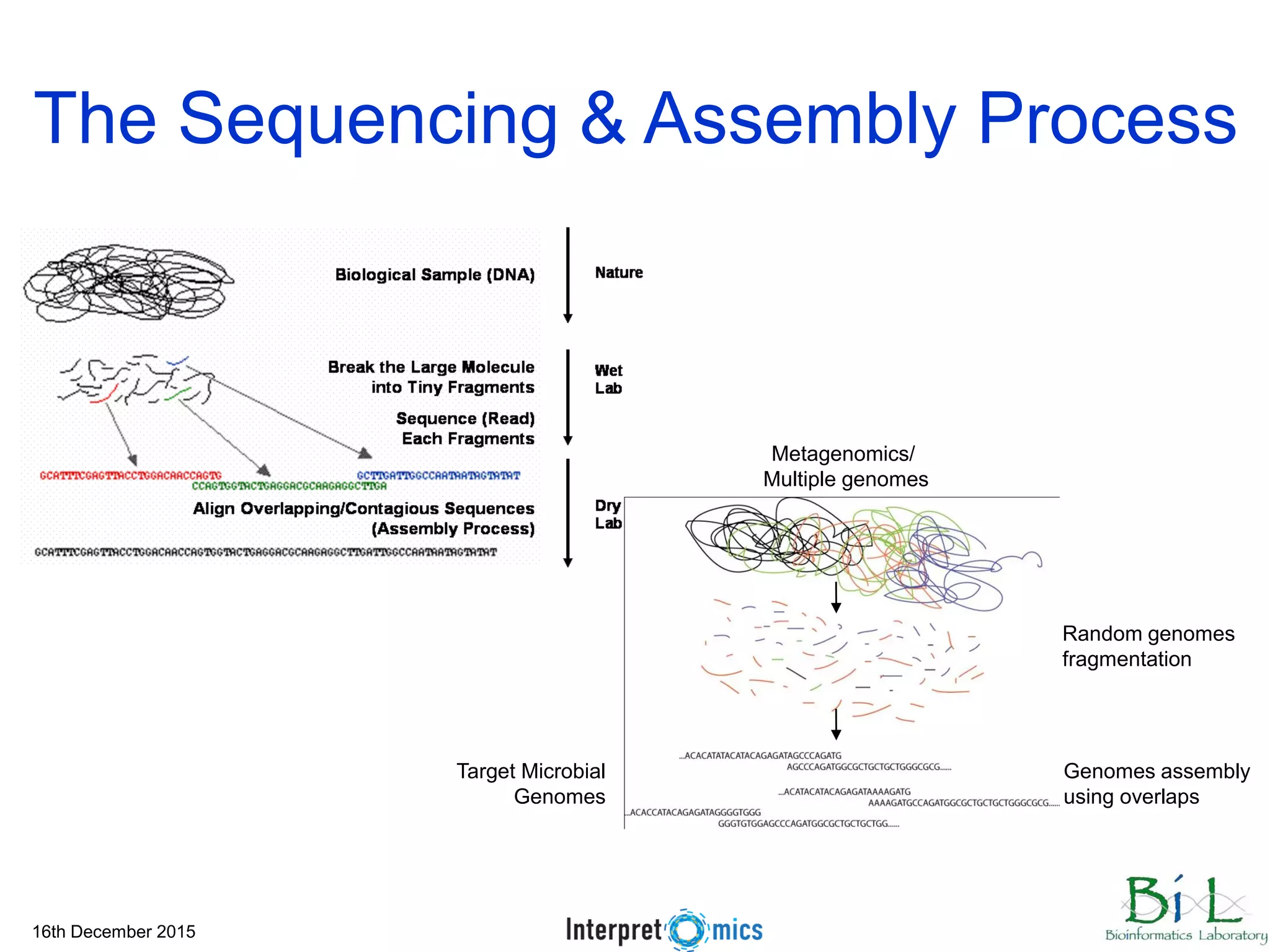






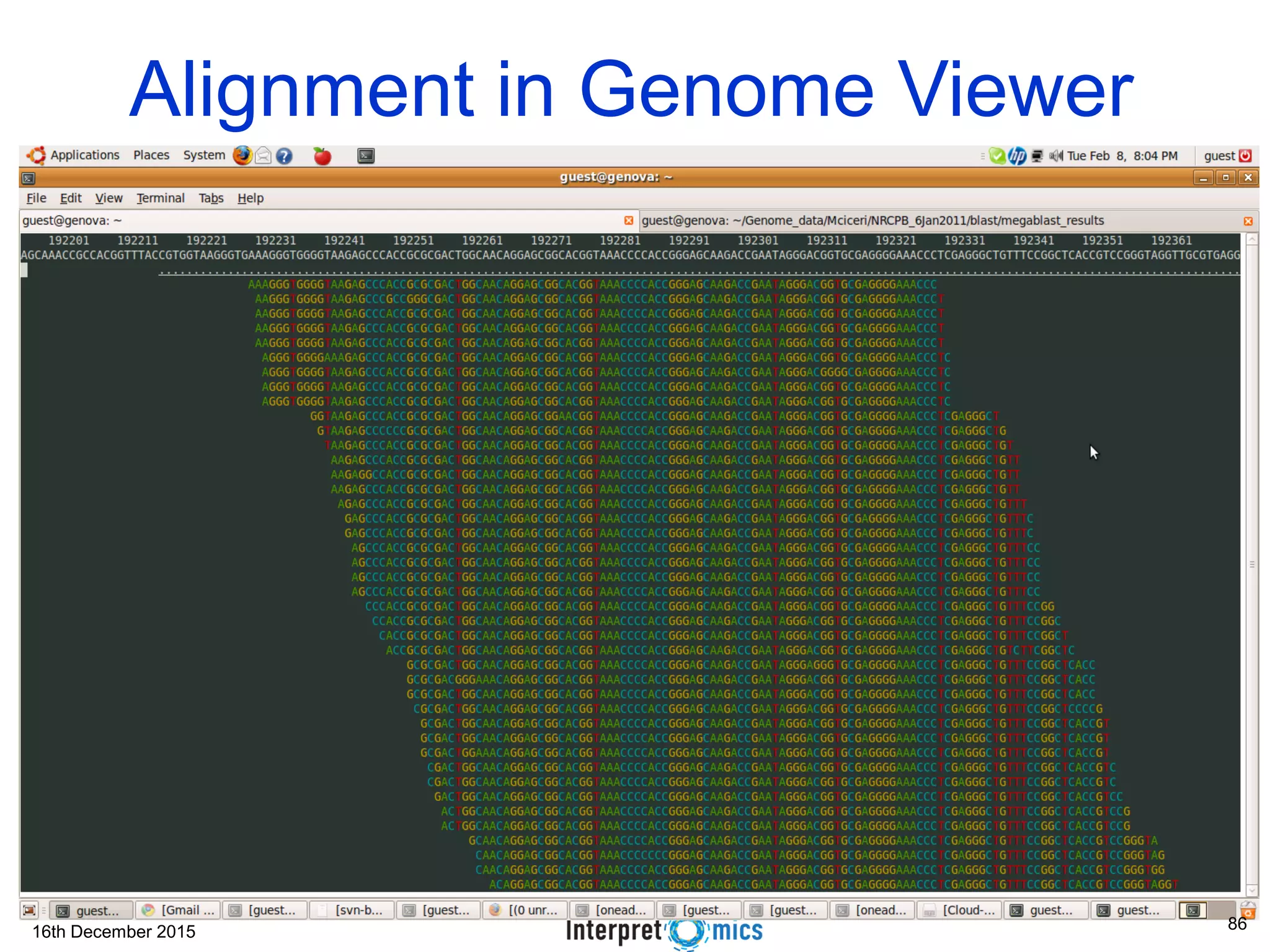




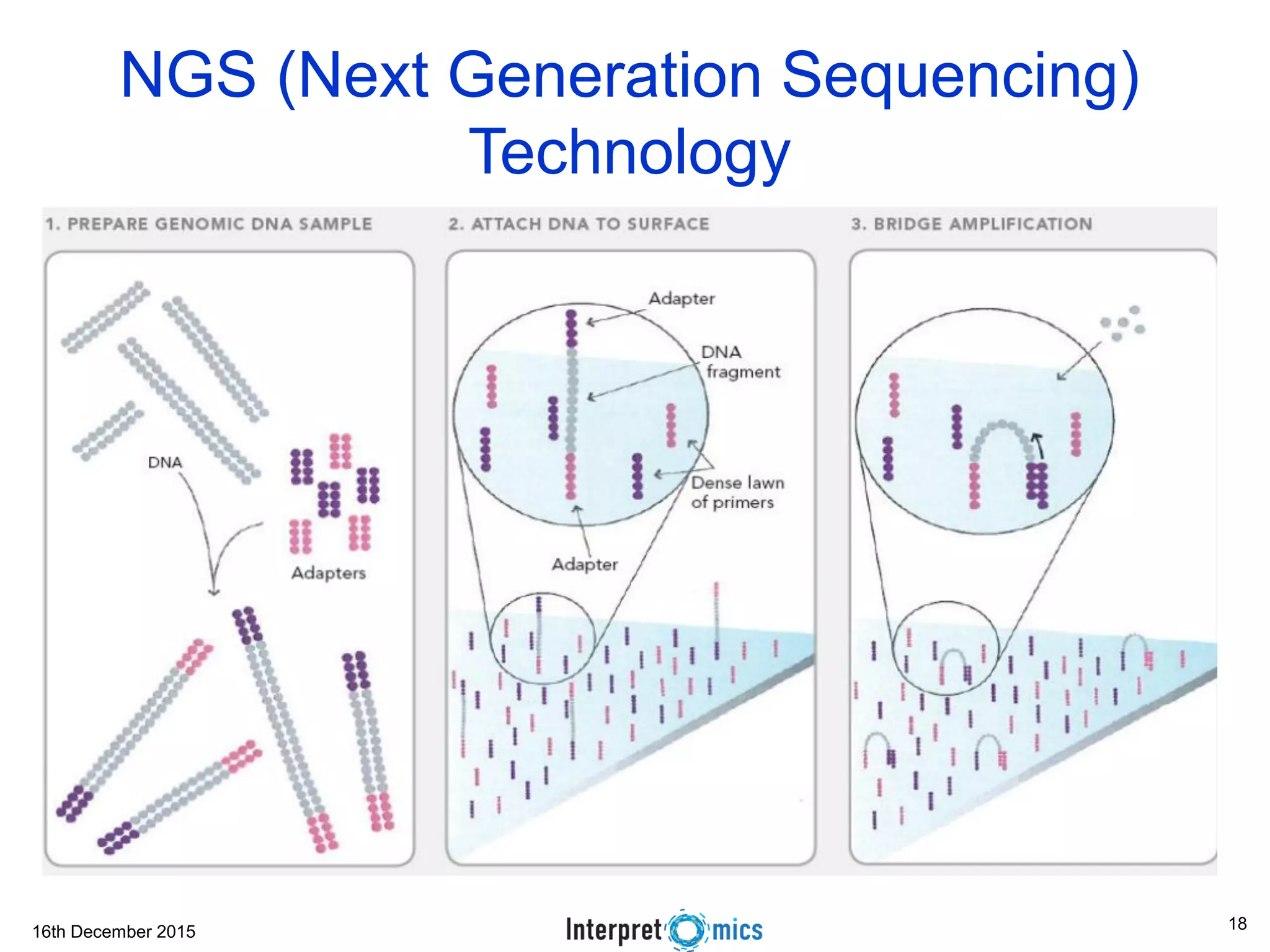
This document summarizes a presentation on genomics and big data in precision medicine. It discusses how next generation sequencing is generating massive amounts of multi-omics data from the genome, epigenome, transcriptome, proteome and metagenome. It describes some of the algorithms and databases used to analyze this big genomic and biological data, including de Bruijn graph algorithms and databases like NCBI, OMIM, and PANTHER. It also discusses some of the challenges in analyzing such large and complex biological data using computational methods.